疾病详情
概论肺间质与间质性肺疾病的概念
肺实质是指各级支气管和肺泡结构。肺间质是指肺内支持组织,包括组成支气管血管周围鞘,小叶间隔和脏层胸膜的结缔组织成分,以及位于肺泡壁之间的间质,即肺泡上皮基底膜与肺毛细血管基底膜之间的间质。肺泡间隔的结缔组织与邻近终末气道、小叶间隔的结缔组织相延续,可达肺门和胸膜。正常肺间质包括细胞成分、细胞外基质(extracellularmatrix,ECM)和结缔组织纤维。细胞成分约占肺间质体积的75%,其中约30%~40%是间质细胞,主要包括成纤维细胞、肌成纤维细胞等,其余是炎症细胞及免疫活性细胞,包括单核巨噬细胞(约占90%)和淋巴细胞(约占10%)。细胞外基质包括基底膜和一些糖蛋白、纤维连接蛋白、层粘连蛋白及其他基质蛋白等。结缔组织纤维包括胶原纤维(约占70%),弹性纤维和网织纤维。
间质性肺疾病(interstitial lung disease,ILD)是一组以肺泡壁和肺泡腔具有不同形式和程度的炎症和纤维化为特征性病理改变,以进行性呼吸困难和X线胸片呈广泛分布的浸润影为主要临床表现的弥漫性肺疾病的总称。因为ILD不仅侵及间质,还累及到肺泡腔、外周气道和血管以及它们各自衬附的上皮和内皮细胞,因此又称为弥漫性实质性肺疾病(diffuse parenchymal lung disease,DPLD)。
间质性肺疾病的分类
间质性肺疾病包括200多种急性和慢性肺部疾病,其中大多数的病因还不明确,已知的原因有职业或环境暴露,药物毒性和结缔组织疾病等,因此临床上通常根据其病因和临床表现对其进行分类(表1)。2002年美国胸科学会/欧洲呼吸学会(ATS/ERS)将其简单分类为:①已知原因如药物、职业或环境暴露、结缔组织疾病等相关的ILD;②特发性间质性肺炎(ⅡP);③肉芽肿性肺疾病如结节病;④以及一组罕见的但具有特征性的临床影像和病理特征的ILD(图1)。


表1 间质性肺疾病的临床分类
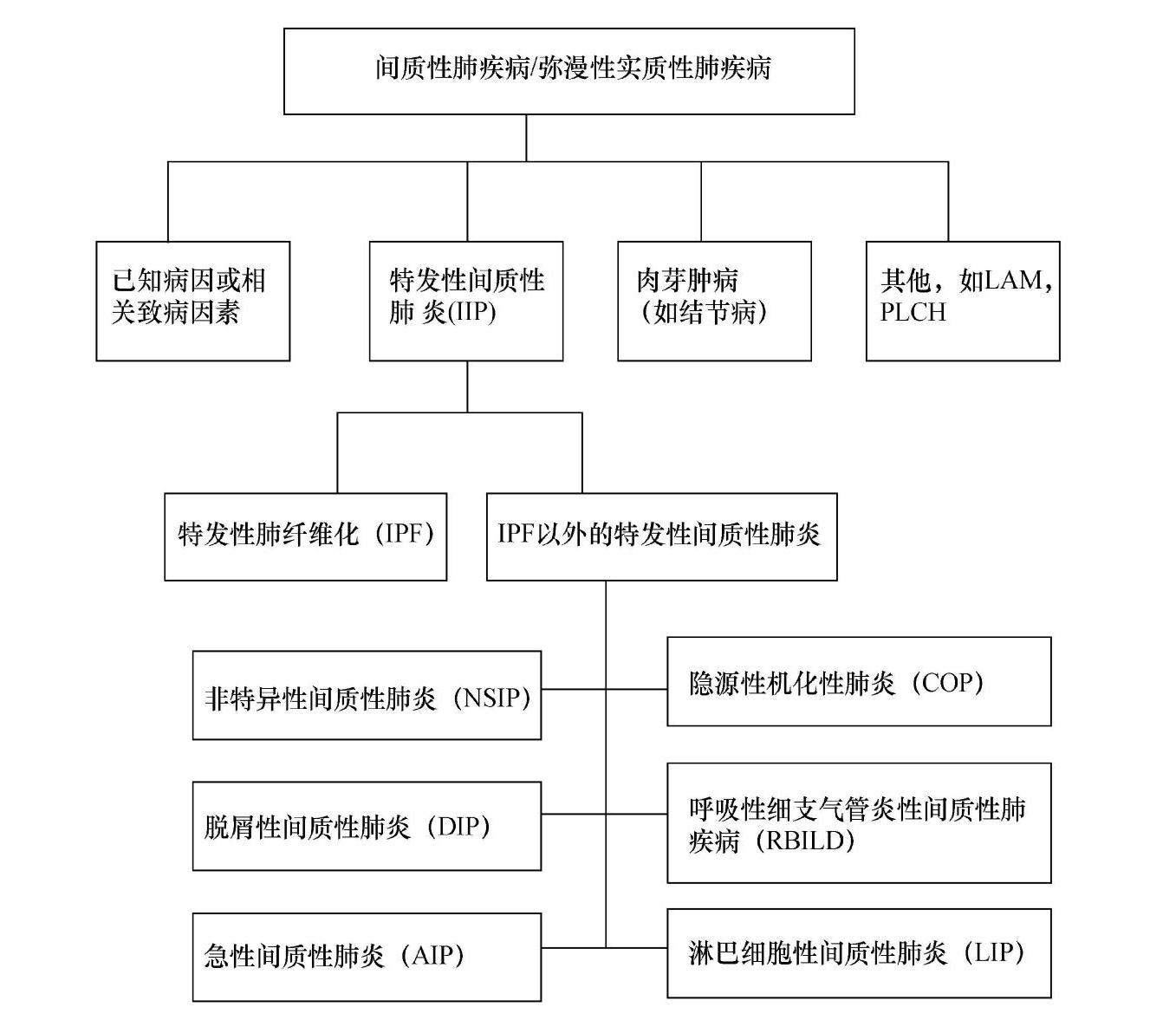
图1 ILD或DPLD的分类(ATS/ERS,2002)
诊断
临床诊断某一种ILD是一个动态的过程,需要临床、放射和病理科医生的密切合作,需要根据所获得的完整资料对先前的诊断进行验证或修订。
(一)临床表现
1.症状
(1)呼吸困难:呼吸困难是ILD患者的最常见和主要症状。多数ILD早期,呼吸困难并不明显,仅在活动时出现,通常不引起重视。而当呼吸困难进行性发展到安静时也出现时,表明病情已经进展到比较严重阶段。
(2)咳嗽:常常表现持续性干咳,尤其常见于那些同时累及气道者。如果咳大量黏液痰要警惕弥漫性支气管肺泡癌的可能。
(3)咯血、喘鸣、胸痛等症状在间质性肺疾病不常见。
(4)全身症状:发热、盗汗、乏力、消瘦、关节肌肉疼痛、肿胀、口眼干燥等肺外表现对于诊断系统疾病如结缔组织疾病等具有提示作用。IPF很少有发热等全身症状。
2.体征
(1)爆裂音或Velcro啰音:两肺可闻及吸气末细小的干性爆裂音或Velcro啰音是ILD的常见体征,尤其是IPF。爆裂音也可出现于胸部影像学正常者。因此,爆裂音对ILD缺乏诊断特异性。
(2)吸气性喘鸣(inspiratory squeaks):有些细支气管炎患者可以在两肺闻及散在的吸气晚期的高调干啰音或吸气性喘鸣。
(3)杵状指:杵状指是ILD患者一个比较常见的晚期征象,通常提示严重的肺结构破坏和肺功能受损,多见于IPF。
(4)肺动脉高压和肺心病的体征:ILD进展到晚期,可以出现肺动脉高压和肺心病,进而表现发绀、呼吸急促、P2亢进、下肢水肿等征象。但是,在一些结缔组织疾病尤其是系统性硬皮病中,肺动脉高压可以为原发性。
(5)关节肿胀或皮肤紧绷可能提示结缔组织疾病。
(二)相关病史
职业或环境接触史详细了解接触物质种类(有机或无机粉尘、化学药物或毒素等)、接触强度、频度和时间;重要的过去病史包括心脏病、过敏性鼻炎和哮喘、结缔组织疾病、肿瘤等;药物应用史,尤其一些ILD相关药物,如胺碘酮、呋喃妥因、博来霉素、甲氨蝶呤等;家族史应注意结节病、IPF等有家族聚集倾向;过敏史包括过敏原、过敏反应表现及转归;吸烟史包括每天吸烟支数、烟龄及戒烟时间;宠物嗜好或接触史,包括鹦鹉或鸽子等的接触强度、频度与时间;居住环境,包括温湿条件、室内装饰、空调或湿化器的应用情况;甚至旅行史。这些病史对于某些ILD具有病因提示作用。
(三)影像学评价
绝大多数ILD患者X线胸片显示弥漫性浸润性阴影。根据疾病累及间质和肺泡的比例不同,胸片或表现以间质浸润为主或表现以肺泡浸润为主。间质病变表现为磨玻璃样变、网格结节或小结节、网格以及蜂窝囊性变,常伴肺容积减小。肺泡浸润呈现边界不清的小结节渗出影,或融合成多发的斑片或片状实变伴支气管气像,或磨玻璃影。
胸部高分辨薄层CT(HRCT)更能细致地显示肺实质异常的程度和性质,能发现X线胸片不能显示的病变。ILD患者在胸部HRCT的影像学表现包括弥漫性结节影、磨玻璃样变、肺泡实变、小叶间隔增厚、胸膜下线、网格状改变、囊腔形成或蜂窝状改变常伴牵拉性支气管扩张或肺结构变形。
影像学检查尤其HRCT应该重点观察病变性质(如磨玻璃样/实变、网状、线条状、结节状、蜂窝或囊腔改变、牵拉性支气管扩张等)、病变分布范围和程度、有无肺门和纵隔淋巴结肿大以及有无胸膜病变。
(四)肺功能
ILD患者以限制性通气功能障碍和气体交换障碍为特征,限制性通气功能障碍表现肺容量包括肺总量(TLC)、肺活量(VC)和残气量(RV)均减少,肺顺应性降低。一秒钟用力呼气容积/用力肺活量(FEV1/FVC)正常或增加。气体交换障碍表现一氧化碳弥散量(DLco)减少,(静息时或运动时)肺泡-动脉氧分压差[p(A-E)O2]增加和低氧血症。DLco是静态参数中最敏感的指标,和肺容量的降低不成比例。运动肺功能试验极其敏感,能够发现轻微或早期的ILD病例,P(A-E)O2于运动后增加更明显。肺功能试验有助于判断肺损害的程度,如果肺功能有严重异常(如VC或TLC低于预计值的60%或DLco低于预计值的40%),2年的病死率将超过50%。
(五)实验室检查
应常规进行血常规和血嗜酸细胞计数、尿常规、肝肾功能、红细胞沉降率(ESR)、抗核抗体(ANA)和类风湿因子(RF)等检查。如果不能除外与结缔组织疾病相关,应测定自身抗体;如怀疑有血管炎,应测定抗中性粒细胞胞浆抗体(anti-neutrophil cytoplasmicantibodies,ANCA)和抗肾小球基底膜抗体;如果不能除外结节病,应测定血清中血管紧张素转化酶(ACE),以及血清钙和尿钙水平;如果病原体感染不能除外,应进行病原体检查尤其是巨细胞病毒(CMV)、肺孢子菌和结核杆菌等;如果肿瘤不能除外,应进行肿瘤相关检查如瘤细胞学检查。总之,这些检查对对ILD的诊断没有特异性,但对ILD的病因或伴随疾病具有提示作用。
(六)支气管镜检查
如果无创性检查不能做出明确诊断,又没有禁忌证,应该进行纤维支气管镜检查并进行经支气管肺活检(transbronchial lung biopsy,TBLB)或/和支气管肺泡灌洗液(bronchoalveolar lavage fluid,BALF)检查,以了解弥漫性肺部渗出性病变的性质。
由于TBLB不能准确取样或取样太小,因此TBLB通常不能对ILD做出确定诊断。然而,对于某些感染性疾病(如结核杆菌、真菌和肺孢子菌感染)、结节病、癌性淋巴管炎、肺泡蛋白沉积症和嗜酸粒细胞性肺炎,多数情况下TBLB能做出确定的诊断。对于ILD患者进行TBLB检查,通常要求进行多次多部位取材至少4~6块,才可能有相对较好的诊断产生率。
BALF检查包括细胞计数与分类,淋巴细胞亚类的分析(表2和表3),还要进行病原体检查和寻找肿瘤细胞。BALF的微生物学和细胞学检查可以提供诊断资料。BAL在非感染性疾病、非恶性肿瘤性间质性肺疾病的诊断和分期中的作用还没有得到证实。然而,许多间质性肺疾病都有特征性的细胞学分类改变,如结节病、外源性过敏性肺泡炎、NSIP和LIP患者的BALF含有明显增多的淋巴细胞,特发性肺纤维化患者的BALF含有明显增多的中性粒细胞或嗜酸粒细胞,嗜酸粒细胞性肺炎患者的BALF中嗜酸粒细胞常大于20%~40%。BALF细胞分类也有助于疾病对治疗的反应和预后判断,淋巴细胞升高者对糖皮质激素的治疗反应好,存活时间长。

表2 BALF的正常细胞学检查结果

表3 BALF的正常淋巴细胞亚类
(七)外科肺活检
外科肺活检包括开胸肺活检(open lung biopsy,OLB)和电视辅助胸腔镜(videoassisted thora-coscopy,VATS)肺活检。胸腔镜肺活检的损伤较开胸肺活检小,能缩短住院时间,因此在大多数病例被优先使用。
如果已经进行的检查(包括TBLB)均无助于建立确定的诊断,如果检查的结果可能改变治疗手段,则需要进行外科肺活检。尤其对于IIP,除了具有典型临床影像表现的IPF病例外,外科肺活检对于确定临床病理诊断是必要的。活检能够将大多数病例确定地分类为已知的病理类型(图2),也能够确定或排除其他诊断,如结节病、淋巴瘤或提示职业病如硬金属疾病。

图2 ILD或DPLD的诊断程序
外科肺活检要求在表现明显炎症和轻度纤维化的病变处取材,要包括邻近病变部位的大体呈正常表现的肺组织,充气后样本组织直径4cm,深度1~1.5cm,避开在表现为严重的纤维化伴有蜂窝肺的病变最严重的部位取材,因为这些部位通常表现为非特异性改变的终末期纤维化。在一个以上的肺叶取材更有帮助。HRCT扫描可以指导外科医生选择最佳的活检部位。
ILD患者的诊断步骤概括如图2:第一步确定是否为ILD,这需要详细的病史、体格检查、X线胸片和肺功能试验。第二步确定是否为IIP,如果有相关的病因如结缔组织疾病、环境因素、药物等可查,则不可能是HP,而是非HP,具体诊断根据相关病因确定。如果无原因可查,则可能是HP,需要做胸部HRCT。第三步进行HRCT诊断,决定是否需要外科肺活检,如果HRCT显示了IPF的特征性改变,结合相应的临床表现,可以建立临床肯定的IPF诊断,不需要进一步检查。如果HRCT显示了其他ILD,如PLCH、PLAM等特征,也可建立临床确定诊断。第四步进行外科肺活检和病理诊断,如果HRCT没有典型的IPF征象,临床表现也不典型,则需要外科肺活检进行病理分型,确定是UIP、NSIP、RB、DIP、DAD、OP和LIP中的一种,还是非IIP。对于一些临床和CT表现都不典型或怀疑其他ILD,可以先进行TBLB或BAL或其他相关试验,如果仍不能确诊,则也需进行外科肺活检,确定病理分型和诊断。
特发性肺纤维化特发性肺纤维化(idiopathic pulmonary fibrosis,IPF)或隐源性纤维化性肺泡炎(cryptogenic fibrosingalveolitis,CFA)是最常见的一种特发性间质性肺炎,是一种原因不明的慢性纤维化性间质性肺炎。它发生于肺,外科肺活检的组织病理学改变是普通型间质性肺炎(UIP)。最近一项关于IPF的流行病学研究资料显示IPF患病率高于以往的统计,且随着年龄的增大而增加,在75岁或75岁以上人群中分别是64.7~227.2/10万人口和27.1~76.4/10万人口。由此,估计美国IPF的患病率和年发病率是14.0~42.7/10万人口和6.8~16.3/10万人口。我国虽然缺乏相应的流行病学资料,但是临床实践中发现近年来IPF的病例呈明显增多的趋势。
病理改变
IPF的病理类型是UIP,UIP型的主要组织学特征是病变主要累及胸膜下外周肺腺泡或小叶并呈斑片状分布。低倍镜下病变呈不均一性,表现为纤维化、蜂窝状改变、间质性炎症和正常肺组织并存。纤维化区显示出时相不均一性,致密的纤维瘢痕区伴散在的成纤维细胞灶。蜂窝状改变是由一些囊性纤维化的气腔组成,这些囊腔通常被覆细支气管上皮并充满黏蛋白。纤维化和蜂窝样变区常见平滑肌增生。间质的炎症常表现肺泡间隔轻度的淋巴细胞和浆细胞浸润,伴Ⅱ型肺泡上皮细胞增生。炎症呈斑片状分布于胶原密集或蜂窝肺区,很少涉及正常的肺泡间隔。
发病机制
目前为止有关IPF的病因还不清楚。由于多半IPF患者有吸烟史,所以吸烟被认为是IPF的潜在危险因素。有几个研究提示了IPF与病毒感染(如EB病毒)的关系,但是并没有研究能证实病毒感染真的就是引起IPF的原因。家族性IPF病例的报道提示IPF存在一定的遗传易感性,但是还没有特定的遗传异常被证实。近来研究发现高达87%~94%的IPF患者都存在胃食管反流,但是两者之间的因果关系还不是十分清楚。其他危险因素包括慢性吸入、药物和环境暴露等。
IPF的发病机制还不十分清楚。过去一直认为不管何种原因引起的纤维化性肺疾病,一个共同机制是炎症损伤肺,刺激纤维形成过度致肺纤维化和结构重建。基于对各型间质性肺炎的病理学知识和在此基础上产生的IPF的定义和IIP的分类,认为IPF的早期是肺泡炎,进而导致肺纤维化形成的这一传统观念已受到挑战。一方面IPF的病理类型是UIP,而在UIP炎症成分很少,几乎没有证据证明IPF开始于炎症过程,或在IPF的早期阶段炎症更明显;另一方面临床研究并没有证实炎症活动标志与IPF的发病阶段和预后的相关性,而且抗炎症反应药物(如糖皮质激素、免疫抑制剂)的治疗效果差。因此,有理由认为炎症启动途径致肺纤维化的机制不是IPF的纤维化形成机制,但它可能代表着非IPF的特发性间质性肺炎的纤维化形成机制,因为它们有一个明显的早期肺泡炎阶段和后期纤维化阶段,早期炎症阶段使用糖皮质激素治疗通常产生良好的反应。
目前认为IPF起源于肺泡上皮反复发生微小损伤后的异常修复。不明原因致肺泡上皮损伤和激活,一方面启动凝血瀑布和组织修复反应;另一方面激活的上皮细胞表达细胞因子和生长因子,启动上皮细胞和成纤维细胞的交互反应,促进成纤维细胞移行到损伤处,并增生和变形为肌成纤维细胞,也自身转分化为肌成纤维细胞,形成成纤维细胞灶。肌成纤维细胞一方面刺激基底膜降解,肺泡上皮细胞凋亡,干扰上皮再生;另一方面增加纤维源性细胞因子,尤其是转化生长因子(TGF)-β的活性以及对它们的反应,产生大量的细胞外基质,也产生金属蛋白酶组织抑制剂TIMP,抑制胶原降解。同时上皮细胞受损,产生金属蛋白酶MMP-1(胶原酶)减少,这种MMPs/TIMPs的失衡使得细胞外基质过度沉积,肺实质结构损伤和纤维化囊腔形成。总之,肺损伤启动了一个复杂的上皮细胞、基质细胞、内皮细胞、炎症细胞、淋巴细胞以及它们释放的各种细胞因子相互作用的过程,最终导致成纤维细胞增生和过量胶原沉积,肺脏正常结构和功能丧失(图3)。
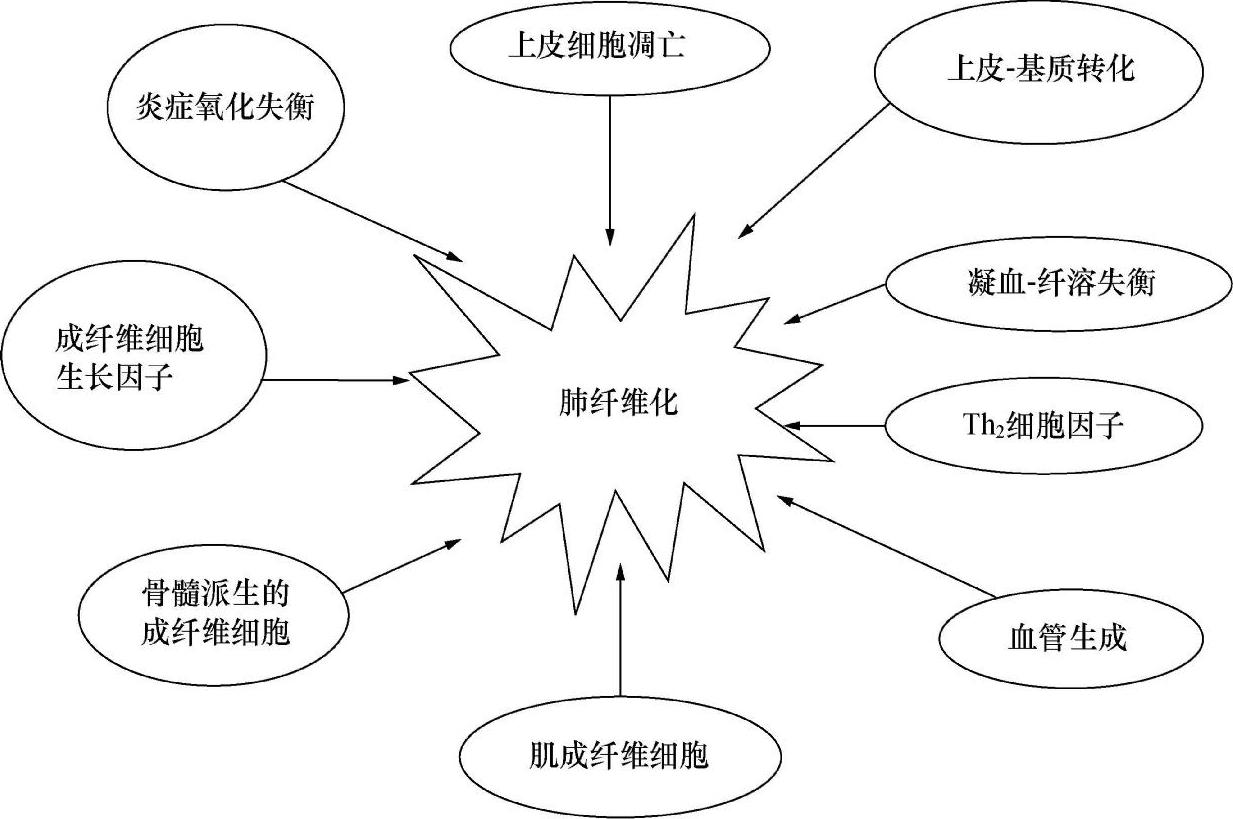
图3 肺纤维化的形成机制
临床表现
1.人口学特征 多于50岁以后发病,男/女为1.5~2/1,75%有吸烟史。
2.症状、体征 呼吸困难是最主要的症状,常表现活动后明显,渐进性加重。其次是干咳。全身症状少见,可以伴有全身不适、乏力和体重减轻等症状,但很少发热。起病隐匿,大部分患者在就诊前6个月已经出现症状。诊断后的中位生存期为2.5~3年。有些患者可以因为急性加重表现呼吸困难加重和肺功能迅速恶化。25%~50%的患者可见杵状指,90%的患者胸部听诊可闻及吸气末细小的Velcro啰音,从双肺基底部,逐渐扩展到整个肺。在疾病晚期可出现明显发绀、肺动脉高压和右心功能不全征象。
实验室及相关检查
(一)影像学改变
IPF患者最常见的胸部影像学异常是双肺外带胸膜下网状或网结节状模糊影,基底部尤为明显,通常伴有蜂窝样变和下叶肺容积减低。5%~10%的IPF患者在首次就诊时的X线胸片表现正常。
IPF在HRCT的特征性表现呈以两肺外带胸膜下和基底部分布为主的斑片性网状或网结节模糊影,常伴有牵拉性支气管扩张和蜂窝肺样改变。无或只有局限性磨玻璃影。罕见胸水或纵隔淋巴结肿大。HRCT诊断UIP的特异性为90%,敏感性为78.5%(图4和彩图5)。
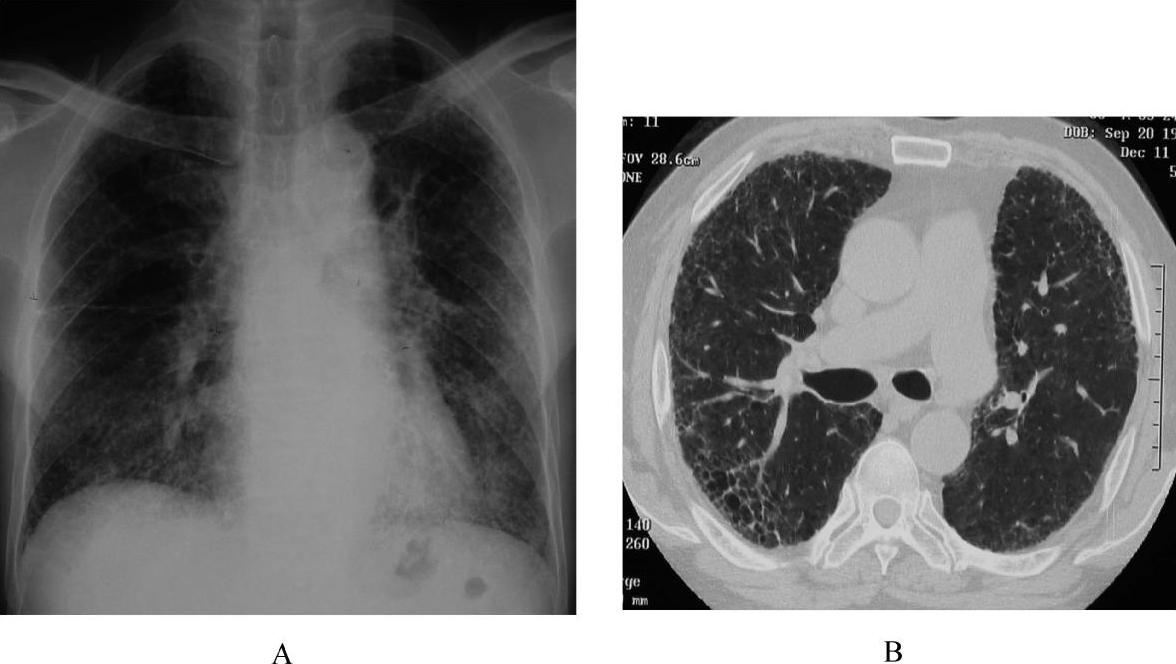
图4 特发性肺纤维化的影像改变
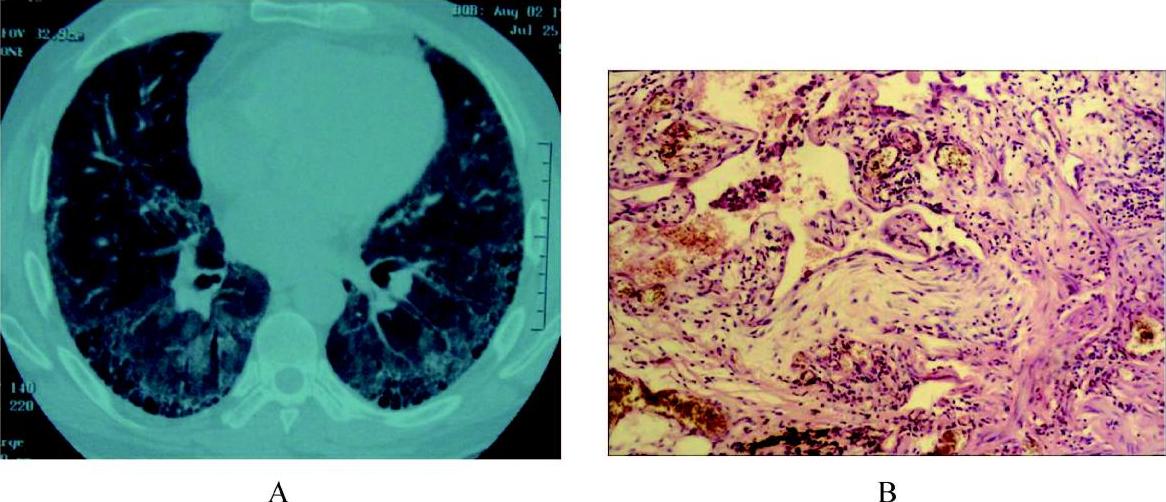
彩图5 特发性肺纤维化的HRCT与病理改变
(二)肺生理功能改变
大部分患者表现为限制性通气障碍,伴DLco减低或DLco/肺泡通气量(VA)降低,静息状态下低氧血症。IPF早期静息肺功能可以正常或接近正常,但运动肺功能表现P(A-a)O2增加和氧分压降低。吸烟和已戒烟的IPF患者与不吸烟的患者相比,因合并慢性阻塞性肺疾病可能导致肺容量相对增高。
(三)血液化验
血液乳酸脱氢酶(LDH)、ESR、抗核抗体和类风湿因子可以轻度增高,但没有特异性。
(四)BALF/TBLB
BALF检查显示中性粒细胞和/或嗜酸性粒细胞增加,淋巴细胞增加不明显。TBLB因为取材太小,不可能做出UIP的病理诊断。因此BAL或TBLB主要的意义在于除外其他疾病,缩小鉴别诊断范围。
(五)外科肺活检
是IPF的主要确诊手段,对于临床或影像学不典型,诊断不清楚,没有手术禁忌证的患者应该考虑外科肺活检。IPF的组织病理类型是UIP(彩图5)。
诊断
(一)有外科肺活检时IPF的诊断
IPF的确定诊断标准包括:①外科取材活检证实病理类型是UIP;②排除其他原因(如环境、药物和结缔组织疾病等)相关的ILD;③限制性肺通气功能障碍与气体交换障碍;④特征性胸片或HRCT改变。
(二)没有外科肺活检时IPF的诊断
如果没有外科肺活检的证据,IPF的临床诊断须同时满足下面4个诊断主要标准和3个次要标准。
主要标准:①排除其他原因(如环境、药物和结缔组织疾病等)相关的ILD;②有限制性肺通气功能障碍和/或气体交换障碍;③HRCT主要显示以两肺外带胸膜下和基底部分布为主的网格状改变伴蜂窝肺,磨玻璃样改变不显著;④BALF或TBLB检查未显示诊断其他疾病的特征(如BALF显示嗜酸性粒细胞或淋巴细胞明显增高,则不支持IPF)。
次要标准:①年龄>50岁;②不能解释的活动性呼吸困难;③隐匿起病,病程>3个月;④两肺基底部吸气时有VelCro啰音。
(三) IPF的急性加重
IPF的急性加重没有统一的诊断标准,多数研究应用的诊断标准是:近30天内的呼吸困难加重;FiO₂/PaO₂<225或PaO₂下降大于10mHg;影像学表现为在网格蜂窝影的基础上出现新的磨玻璃样异常;可除外其他疾病,如感染、左心衰竭或肺栓塞等。最近,由美国国立卫生研究院资助的“IPF临床研究的国际合作项目”(IPFnet)的项目组在总结关于IPF急性加重的研究成果和经验的基础上,提出了新的诊断标准如表4。

表4 IPF急性加重的诊断
鉴别诊断
IPF的鉴别诊断首先通过病史、临床表现和相应检查和其他原因或疾病相关的ILD相鉴别。进一步还需要和其他类型的IIP进行鉴别(表5)。
(一)非特异性间质性肺炎非特异性间质性肺炎(nonspecific interstitial pneumonia,NSIP)是肺活检组织学特征不符合UIP、OP、DAD、DIP或LIP的一种间质性肺炎类型。根据病理表现分为细胞型NSIP和纤维化型NSIP,或两者兼而有之,病变时相均一,弥散。
特发性NSIP是IIP的常见类型,平均发病年龄52岁(26~73岁),男/女约1∶2,慢性或亚急性起病,主要症状是呼吸困难(96%)和咳嗽(87%),其他如体重减轻25%,发热22%,关节痛14%,关节炎3%,杵状指14%。胸部CT/HRCT主要表现双下肺分布为主的斑片或片状磨玻璃样变,常伴网格样变,还可以伴牵拉性支气管扩张或蜂窝状改变,多见于纤维化型。支气管肺泡灌洗液的特点与UIP不同,大约50%的病例淋巴细胞比例增高。对糖皮质激素的反应和预后较好,5年生存率82.3%,10年生存率73.2%。
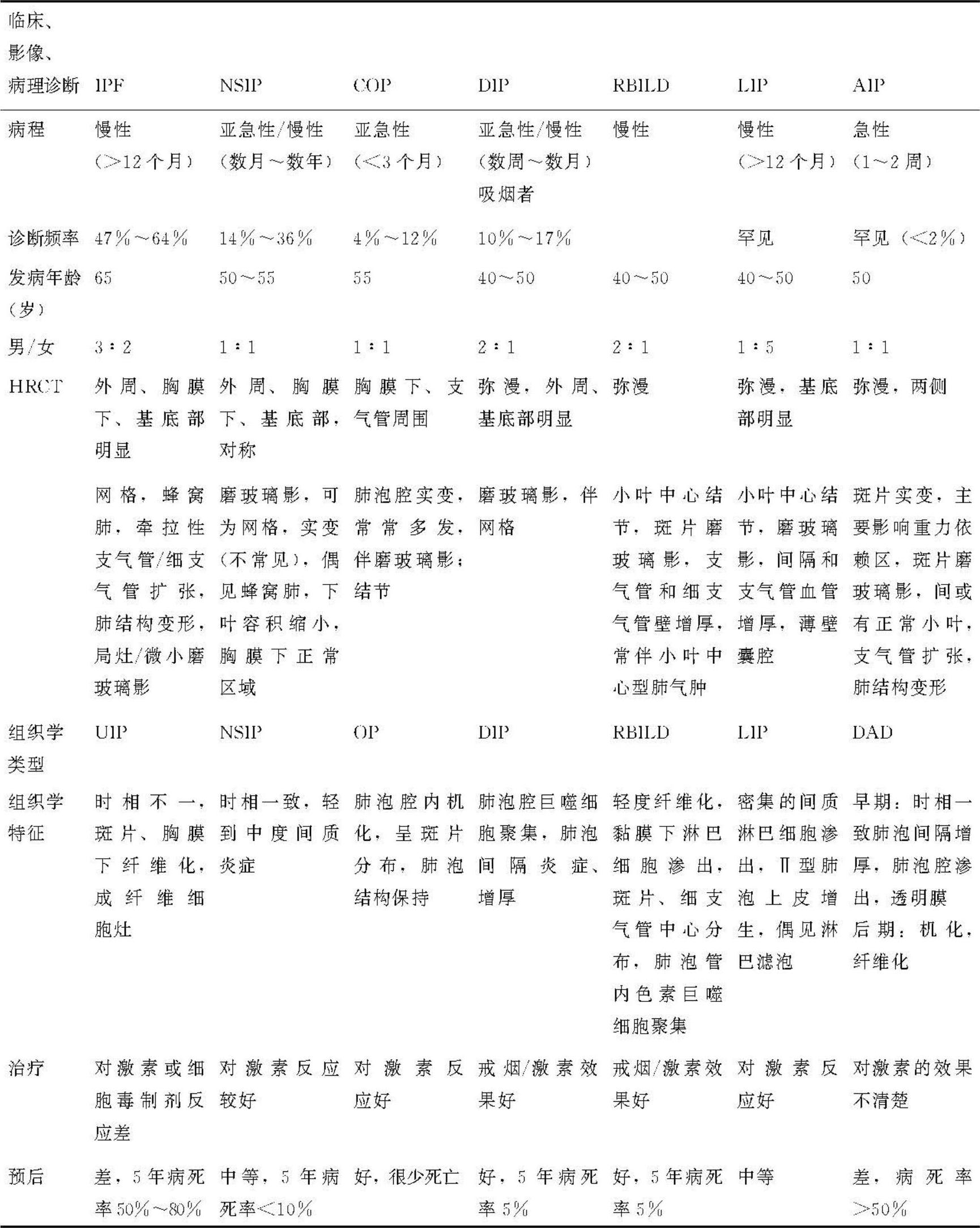
表5 特发性间质性肺炎的临床、影像、病理及预后比较
(二)隐源性机化性肺炎
机化性肺炎是一种与多种病因相关的组织学类型。原因不明的机化性肺炎称为隐源性机化性肺炎(cryptogenicorganizing pneumonia,COP)或特发性闭塞性细支气管炎伴机化性肺炎(bronchiolitis obliteransorganizing pneumonia,BOOP)。病变呈小叶中心分布的斑片性表现,以肺泡管和肺泡腔内机化性肉芽组织栓子形成伴或不伴细支气管腔内息肉为特征。病变时相一致,背景的肺泡结构相对完整。
平均发病年龄55岁,多数亚急性起病,以呼吸困难或活动后气短伴持续性干咳或咳白痰为主要症状。发病前1~3个月多有类似亚急性呼吸道感染的前驱症状,如咳嗽、发热、寒战、出汗、全身不适、乏力和体重减轻。肺部听诊常表现局限性或较广泛的吸气时爆裂音,极少发现实变征象。杵状指罕见。常见红细胞沉降率、C反应蛋白和外周血中性粒细胞显著增加。胸部CT/HRCT为多发的斑片或片状实变影,实变区见支气管气像或轻度柱状支气管扩张。磨玻璃影见于大约60%的病例,通常与肺实变相伴随。病变可以游走。BALF检查常见淋巴细胞总数和比例增加(淋巴细胞的比值可高达40%),CD4/CD8比值减低。COP对糖皮质激素的治疗反应好,但合适的剂量和疗程尚不清楚。通常应用泼尼松,起始剂量0.75~1mg/(kg·d),依据治疗反应逐渐减量,疗程6~12个月。COP容易复发,但复发COP对激素治疗依然敏感。COP的预后好,中位存活10年以上,病死率为0~12%。
(三)急性间质性肺炎
急性间质性肺炎(AIP),即Hamman-Rich综合征(Hamman-Rich Syndrome),是一种发病原因未明的病情进展迅速的间质性肺炎。通常表现既往健康个体,于几天到几周内出现迅速发展的呼吸衰竭,病理呈弥漫性肺泡损伤(DAD)的急性和/或机化期。它的临床表现、影像学、病理生理及病理改变特点极类似感染中毒症和休克所致的急性呼吸窘迫综合征(ARDS),因此AIP也被认为是一种原因不明的ARDS,即特发性ARDS。对AIP目前尚无确切有效的治疗方法,病死率高达50%以上。
(四)呼吸性细支气管炎相关性间质性肺疾病
呼吸性细支气管炎相关性间质性肺疾病(respiratory bronchiolitis interstitial lung disease,RBILD)是与呼吸性细支气管炎病理学损伤有关的间质性肺疾病的临床诊断,以终未细支气管、呼吸性细支气管和邻近的肺泡腔聚集大量棕黄色色素沉着的巨噬细胞为特征,伴肺泡间隔轻度增宽或细支管周围轻微纤维化。
RBILD通常发生于40~50岁吸烟者,男/女≈2/1,多仅有轻微咳嗽和呼吸困难,30%有爆裂音,杵状指罕见。胸部X线检查发现支气管肺纹理增重,或伴磨玻璃影。胸部CT/HRCT表现弥漫分布的细支气管中心性结节,磨玻璃影与过度充气的肺泡构成马赛克征。可见中央与外周气道的管壁增厚。肺功能通常显示DLco降低,气流轻度阻塞,肺容量正常。BALF以包含色素沉着的肺泡巨噬细胞为特征。某些患者经过戒烟和/或糖皮质激素治疗后,RBILD的CT表现可以恢复正常。因此,首选戒烟治疗,对症状明显的患者采用糖皮质激素治疗。
(五)脱屑性间质性肺炎
脱屑性间质性肺炎(desquamative interstitial pneumonia,DIP)是以弥漫均一的肺泡腔内大量巨噬细胞聚集为主要病变特点,伴轻或中度肺泡间隔增宽,很少形成成纤维细胞灶和蜂窝肺改变。由于与RBILD的病理学改变相似,绝大多数与吸烟有联系,因此许多人认为这种疾病代表RBILD的终末期。
DIP常于40~50岁的吸烟者中发病,男/女≈2/1,隐匿起病,主要表现呼吸困难或伴咳嗽,60%有爆裂音,40%有杵状指。胸部X线/CT表现主要是两肺基底部,胸膜下分布明显的磨玻璃变或伴网格影,与IPF的区别是磨玻璃样变突出,无蜂窝肺样改变。肺功能主要表现为限制性通气功能障碍,肺弥散功能障碍及低氧血症。BALF同RBILD,以含色素颗粒沉着的肺泡巨噬细胞增加为特征。DIP的预后总体较好,多数患者在戒烟和应用糖皮质激素治疗后病情缓解,10年生存率约为70%。
(六)淋巴细胞性间质性肺炎
淋巴细胞性间质性肺炎(lymphocytic interstitial pneumonia,LIP)定义为致密的间质淋巴浸润,包括淋巴细胞、浆细胞和组织细胞,伴Ⅱ型细胞增生和肺泡巨噬细胞轻度增加。LIP常与一些淋巴增殖性疾病如淋巴瘤,结缔组织疾病和免疫缺陷引起的继发性淋巴增殖有关,真正的特发性LIP发病率很低。特发性LIP的临床表现也很不清楚。多见于女性,可以出现在任何年龄段,但常于50岁时被诊断。起病缓慢,常于3年或更长的时间内出现逐渐加重的呼吸困难和咳嗽,全身症状罕见。特发性LIP极少进展为纤维化,没有杵状指、爆裂音以及IIP的生理学特征或表现轻微。胸片表现肺部浸润影主要分布在基底部和呈弥漫性改变伴蜂窝影。CT主要表现为磨玻璃影,或伴网格状影。BALF检查可见大量淋巴细胞。一旦LIP型的组织学诊断成立,临床医生应重新评价患者,是否具有与LIP相关的潜在疾病。糖皮质激素是使用最为广泛的药物,经过治疗大部分患者症状消失或改善,但对治疗是否会影响疾病的进程或显著影响肺生理学尚不清楚。偶尔可见自然缓解的病例。
治疗
1.对症支持治疗 IPF至今没有有效的治疗方法。康复治疗、心理治疗以及针对低氧血症的氧疗等对于树立患者信心、减轻患者症状,改善生活质量具有非常重要的意义。
2.糖皮质激素联合细胞毒性制剂 ATS/ERS以及中华医学会呼吸分会推荐方案如下:①糖皮质激素+硫唑嘌呤或;②糖皮质激素+环磷酰胺。泼尼松初始剂量为0.5mg/(kg·d),连续4周;第5周开始减量为0.25mg/(kg·d),连续8周;第13周减量为0.125mg/(kg·d),并维持治疗。硫唑嘌呤或环磷酰胺口服剂量为2mg/(kg·d),最大量150mg/d。通常以25~50mg为起始剂量,每1~2周增加25mg,直至每日最大剂量。治疗过程中,每3个月复查一次,如果没有严重并发症或副反应,联合治疗时间不应短于6个月。治疗6~12个月后,如果病情改善或稳定,则继续联合治疗。如果病情加重,应该停药或改用、合用其他药物。治疗满18个月后,是否继续治疗需根据临床反应和病人的耐受性而做决定。
疗效以符合下列2个或2个以上条件者为有效,否则无效:①症状减轻(咳嗽、呼吸困难);②X线胸片/HRCT显示肺间质病变减轻;③肺功能改善(TLC或VC增加、DLco增加、PaO2或SaO2增加)。
然而,迄今为止没有研究证实该治疗方案能改善IPF患者的生存和预后,因此新近发表的英国胸科学会(BTS)指南已经不作推荐,但是认为在采用ATS/ERS建议的泼尼松与硫唑嘌呤联合治疗方案,或更小剂量的泼尼松(≤20mg/d)和/或硫唑嘌呤联合应用的基础上,同时应用N-乙酰半胱氨酸1800mg/d,分3次口服的方案是可行的。
3.新型抗纤维化制剂 虽然干扰素(interferon)、吡非尼酮(pirfenidone)、内皮素受体抑制剂如波生坦(bosentan),TNF-α抑制剂或受体拮抗剂如依那西普(etanercept)等新型药物具有潜在的治疗作用,但还没有足够的临床试验证据证明这些药物可以改善IPF患者的生存与预后。鉴于IPF的治疗困难及新药的可能治疗作用,因此BTS指南建议如果可能,应推荐确诊的IPF患者参与高质量的临床治疗试验。
4.肺移植 目前IPF肺移植的5年存活率超过50%,终末期IPF的惟一有效治疗方法是肺移植。因此,如果可能应该积极推荐确诊的IPF患者考虑肺移植。IPF患者的肺移植指征:组织病理学或放射影像学符合UIP的改变,具有下列任一表现者,DLco<39%预计值;随访6个月内FVC下降幅度大于10%或DLco下降大于15%;6分钟步行试验SpO2小于88%;HRCT的纤维化评分大于2。当组织学证实有NSIP改变时,符合下列任一条件者,DLco<35%预计值;6个月内FVC下降幅度大于10%或更大,DLco下降15%。
结节病结节病(sarcoidosis)是一种原因不明的多系统肉芽肿性疾病,主要侵犯肺和淋巴系统,其次是眼部和皮肤。
流行病学
结节病常见于中欧、美国和日本,少见于中美洲和南美洲、日本以外的亚洲国家及非洲。患病率从不足1/100000到高于50/100000都有报道,以斯堪的那维亚国家和美籍非洲人群的患病率最高。我国对结节病的认识相对较晚,自1958年报道首例以来,1982年中华结核和呼吸系统疾病杂志编委会综合报道129例,近10年来随着我国对结节病的认识水平提高,结节病已经不再像过去那样少见。
结节病多发于中青年(<40岁),发病高峰年龄在20~29岁间,女性发病稍高于男性。斯堪的那维亚国家,德国和日本女性有第二个高峰,发病年龄大于50岁。结节病在美籍非洲人表现常常严重,而在白人常常无症状。结节性红斑通常是急性结节病和预后良好的表现,见于18%的芬兰和30%的英国结节病患者,很少见于美籍非洲人和日本人。相反,冻疮样狼疮是结节病的慢性表现,美籍非洲人较白人似乎有更高的出现率。ACCESS(a case-control etiologic studyof sarcoidosis)研究发现肺外淋巴结肿大,肝、眼、皮肤(结节性红斑除外)、骨髓受累多见于美籍非洲人。异常钙代谢多见于白人,年龄大于50岁。结节性红斑更多见于妇女,无明显种族差异。胸外淋巴结肿大多见于小于40岁患者。
病因与发病机制
1.遗传因素
(1)家族遗传:结节病具有一定的家族聚集性。ACCESS是迄今为止病例数最多的一项探讨结节病病因的病例对照研究,来自美国的种族、性别、年龄±5岁和地区匹配的706对结节病和对照病例有10862例一级亲属和17047例二级亲属。结果显示家族中结节病在兄弟姐妹发病的相对危险度最高(OR=5.8,95%CI=2.1~15.9),高于父母亲(OR=3.8,95%CI=1.2~11.3)。结节病患者的一级亲属(兄妹或父母)中结节病的病例数比对照组高5倍。结节病患者的一级亲属和二级亲属的患病危险度增加提示遗传因素在结节病发病中的重要作用。(2)易感基因:人类白细胞相关抗原(HLA)表型与结节病的关系研究揭示遗传易感性可以解释不同人种和种族在临床表现和严重程度的异质性。在捷克和意大利,结节病的急性发病(包括Löfgran综合征)和预后良好与HLA-A1/B8/CW7/DR3等位基因有关。在斯堪的那维亚,良好的预后以及较短的病程与HLA-DR17有关;相反,病程拖延与DR14和DR15有关。慢性结节病在日本人中与B13有关,在非洲裔美国人与BW15有关。结节病与第6染色体、HLA区域和CCR5化学趋化因子受体基因多态性有关,与ACE基因多态性关系不明确,而与TNF-a、TGF-β或IL-10基因表达形式无关。最近的研究发现维生素D受体基因多态性及B等位基因是结节病发病的遗传危险因素。总之,结节病的临床表型以及患病的种族差异提示有遗传因素的作用,家族和病例对照研究证实了结节病的遗传易感性。系列结节病的遗传易感基因研究证实与结节病表型关系最为密切的基因位于6号染色体的MHC区域。其他候选基因如细胞因子、化学趋化因子受体等均不具备可重复性,功能的有效性未能得到证实。因此,需要进一步开展结节病(包括家族结节病)的队列研究、精确定义结节病的临床表型以及发展新的技术以描绘出结节病的临床表型和遗传易感性的关系图谱。
2.环境因素 一些病原体如EB病毒、伯氏疏螺旋体(Borrelia burgdorferi)、痤疮丙酸杆菌(Propionibacterium acne)、结核和其他分枝杆菌等作为结节病的可能病因没有被证实。至今为止没有感染性病因或其他因素被一直证明与结节病发病的关系。
3.免疫机制 结节病以受累脏器,尤其是肺的非干酪样坏死性肉芽肿为病理特点,病变组织聚集大量激活的Thl型CD4+T细胞和巨噬细胞是其特征性免疫异常表现,这些细胞及其产生的细胞因子和其他介质共同促进肉芽肿形成。导致结节病肉芽肿形成的始动因素尚不明确。激活的肺泡巨噬细胞识别、加工、呈递结节病相关的抗原到Thl细胞,同时也释放白介素-12(IL-12)和IL-18,IL-12和IL-18诱导Thl型免疫反应并刺激肺T细胞激活,后者表达IL-2受体和HLA-DR,并产生干扰素-y(IFN-γ)和IL-2,促进单核细胞/巨噬细胞进一步聚集在疾病活动部位。IFN-γ激活巨噬细胞,促进巨噬细胞转化成巨细胞。肺泡巨噬细胞增加前炎性细胞因子IL-1、IL-6和TNF-α及化学趋化因子如RANTES(regulatedonactivation,normal T cellexpressedand secreted)的释放,促进肉芽肿形成和维持。
激活的巨噬细胞能够分泌各种纤维源性因子如转化生长因子(TGF-β)、血小板派生的生长因子(PDGF)、胰岛素样生长因子-1(IGF-1),促进成纤维细胞的增生、胶原的合成以及纤维化的形成。目前尚无研究能说明为什么仅有少数结节病患者肺部病变持续,并导致纤维化。从Th1型向Th2型的转换可能是疾病持续进展的重要因素。从Th1转换到Th2免疫反应,Th2细胞释放IL-4刺激细胞外基质蛋白和化学趋化因子产生,促进成纤维细胞形成和肺纤维化。
虽然结节病的确切病因和发病机制尚不清楚,有待进一步研究,但是目前认为结节病是由于遗传易感者受特定的环境暴露刺激,导致受累脏器局部产生Th1型免疫反应所致。
病理
结节病的特征性病理改变是非干酪样上皮样细胞性肉芽肿,主要由高分化的单核吞噬细胞(上皮样细胞和巨细胞)和淋巴细胞组成。巨细胞可以有包涵体如舒曼小体(Schaumanbodies)和星状小体(asteroid bodies)。肉芽肿的中心主要是CD4+淋巴细胞,而外周主要是CD8+淋巴细胞。结节病性肉芽肿结局或消散,或发展成纤维化,纤维化通常始于外周,向中心进展,导致完全纤维化和/或透明样变。结节病肉芽肿经常累及淋巴结(尤其胸内淋巴结)、肺、肝、脾和皮肤。在肺75%的肉芽肿沿淋巴管分布,接近或位于支气管血管鞘、胸膜下或小叶间隔,开胸肺活检或尸检发现半数以上累及血管。终末期结节病导致肺实质纤维化和蜂窝肺。
临床表现
结节病的临床过程表现多样,与起病的急缓和器官受累的不同以及肉芽肿的活动性有关,还与种族和地区有关。
(一)急性结节病(Löfgren's syndrome)
表现为双肺门淋巴结肿大、关节炎和结节性红斑,常伴有发热、肌肉痛、不适。85%的患者于一年内自然缓解。
(二)亚急性/慢性结节病
约50%的结节病无症状,体检或胸片偶尔发现。
1.系统症状 约1/3患者可以有非特异性表现,如发热、体重减轻、无力、不适和盗汗。
2.胸内结节病 90%以上的结节病可累及肺,其临床表现比较隐匿。30%~50%会出现咳嗽、胸痛或呼吸困难,罕见咯血。20%会出现气道高反应性或伴喘鸣音,不足20%有爆裂音,杵状指罕见。罕见胸水、胸膜增厚或钙化以及乳糜胸或气胸。
3.胸外结节病
(1)淋巴结:在30%~40%的患者能触及淋巴结肿大。最常受累的是颈、腋窝、肱骨内上髁、腹股沟淋巴结。肿大的淋巴结分散,可活动,无触痛,不形成溃疡和窦道。
(2)皮肤:25%的结节病累及皮肤,表现为皮肤结节性红斑(多位于下肢伸侧,6~8周内消散)、冻疮样狼疮(lupus pernio)和皮下结节等(彩图6a)。
(3)眼:11%~83%累及眼部,以葡萄膜炎最常见,急性前葡萄膜炎可自然缓解或糖皮质激素局部治疗缓解,慢性葡萄膜炎致青光眼、白内障和视力消失。其他眼部病变包括结膜滤泡、泪腺增大、角结膜干燥、泪囊炎和视网膜血管炎。
(4)心脏:尸检发现30%累及心脏,但临床只发现5%,主要表现为心律失常、心脏衰竭或猝死。
(5)肝:活检发现50%~80%累及肝,但临床只发现20%有肝大,30%有血清碱性磷酸酶和转氨酶增高。
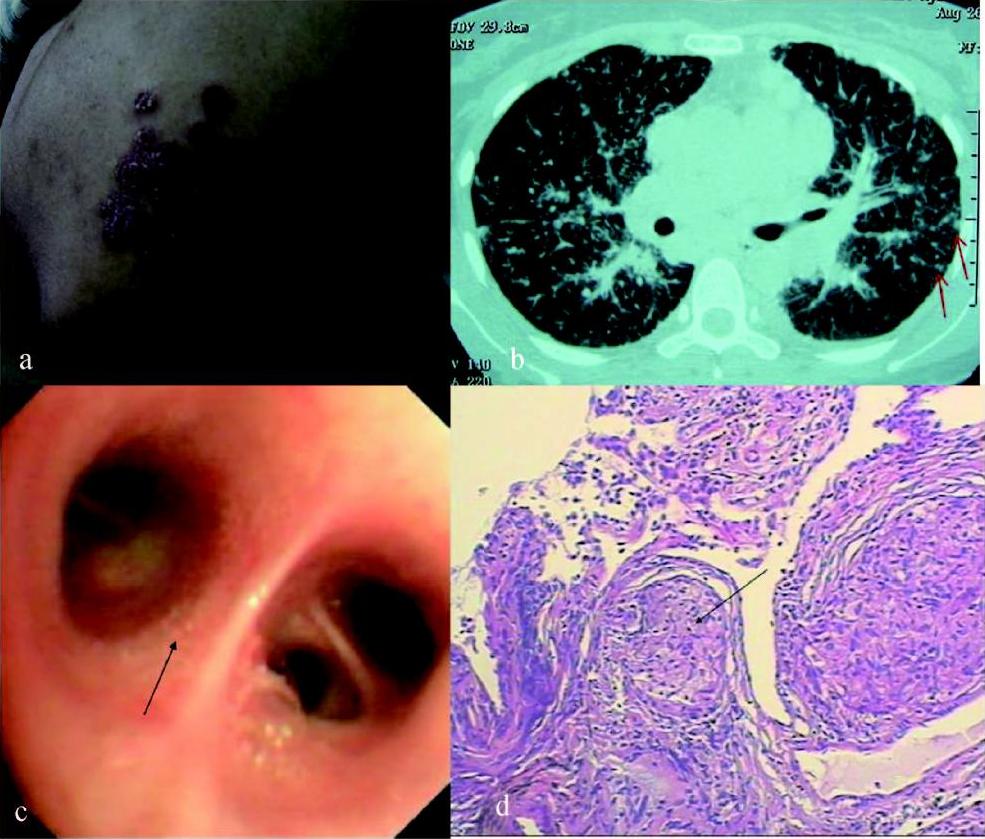
彩图6 女性患者,36岁,皮肤结节16个月,咳嗽、活动 后气短8个月,诊断结节病
(6)肌肉骨骼系统:25%~39%有关节疼痛,但关节畸形少见。关节受累可以是急性一过性,也可以为慢性持续性,肌肉受累并出现症状的较少。慢性肌病多发生于妇女,并可能是唯一表现。必要的滑液或肌肉活检可以发现非干酪性肉芽肿。骨囊肿只在有慢性皮肤病变时才出现。
(7)神经系统:临床上可识别有神经系统受累不足10%,以脑基底部最常见。常见面神经麻痹、下丘脑和垂体病变。
(8)内分泌:2%~10%有高钙血症.高尿钙的发生率大约是其3倍。高钙血症与激活的巨噬细胞和肉芽肿使1,25(OH)2D3的产生调节障碍有关。持续的高血钙和高尿钙可以引起肾钙化、肾衰竭。
(9)其他系统:腮腺、胃肠道、血液、肾以及生殖系统等也可受累。
辅助检查
(一)影像学检查
1.胸部X线检查 90%以上的患者表现X线胸片异常,胸片是提示诊断的敏感工具,双侧肺门淋巴结肿大(BHL)(具有或不具有右侧气管旁淋巴结肿大)是最常见的征象(图7)。临床上通常根据后前位X线胸片对结节病进行分期(表6),目前对这种分期尚存在争议。

表6 结节病的胸部X线分期
2.胸部CT/HRCT HRCT的典型表现为沿着支气管血管束分布的微小结节和融合成球的肺泡渗出。其他异常表现有磨玻璃样变、索条带状影、蜂窝肺、牵拉性支气管扩张以及血管或支气管的扭曲或变形。病变多侵犯上叶,肺底部相对正常。可见气管前、气管旁、主动脉旁和隆突下区的淋巴结肿大(彩图6b)。
3.67Ga核素扫描 肉芽肿活性巨噬细胞摄取67Ga明显增加,肉芽肿性病变可被67Ga显示,除显示Panda和Lamba图像具有诊断意义外,通常无诊断特异性,但可以帮助判断结节病的活动性。
(二)肺功能试验
80%以上的I期结节病患者的肺功能正常。Ⅱ期或Ⅲ期结节病的肺功能异常者占40%~70%,特征性变化是限制性通气功能障碍和弥散量降低及氧合障碍。约1/3以上的病人同时有气道阻塞。

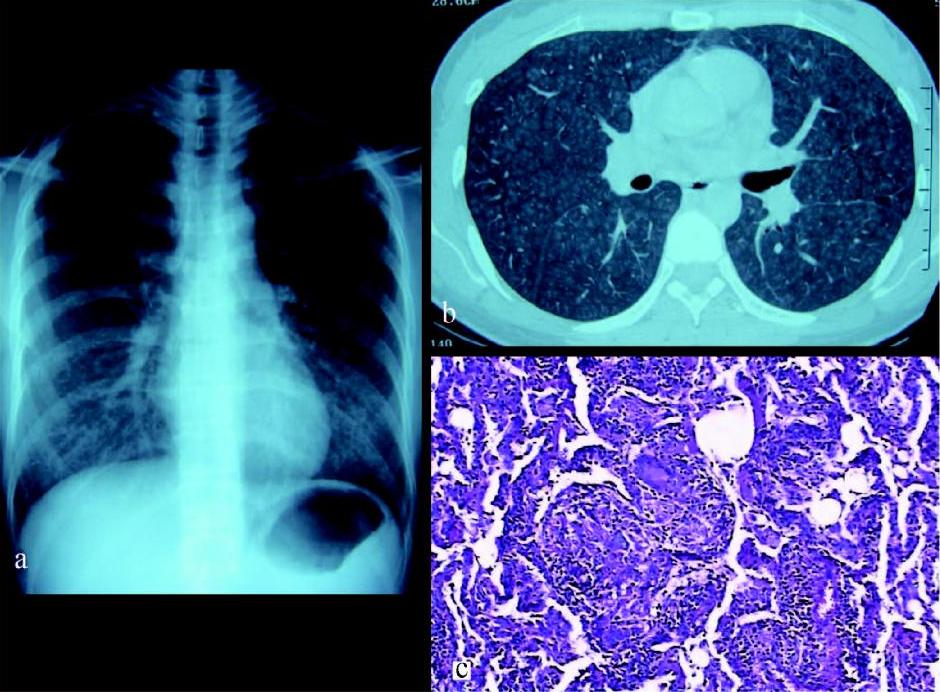
图7 36岁患者,体检胸片发现双侧 肺门淋巴结肿大,诊断结节病I期
(三)纤维支气管镜与支气管肺泡灌洗
支气管镜下可以见到因隆突下淋巴结肿大所致的气管隆突增宽,气管和支气管黏膜受累所致的黏膜结节(彩图6c)。BALF检查主要显示淋巴细胞增加,CD4+/CD8+的比值增加(>3.5)。结节病可以通过纤维支气管镜黏膜活检,经支气管肺活检(transbronchial lung biopsies,TBLB),经支气管淋巴结针吸活检(transbronchial needle aspiration biopsies,TBNA)得到诊断,这些检查的诊断产生率较高,风险很低,成为目前肺结节病的重要确诊手段,尤其是TBLB(彩图6d)。一般不需要纵隔镜或外科肺活检,除非经支气管镜活检或BAL也无法诊断。
(四)其他实验室检查
1.常规实验室检查 约1/3患者有轻度贫血和全血细胞减少。2%~10%患者有高钙血症,30%患者有高尿钙症。另外,血清γ-球蛋白、C反应蛋白、红细胞沉降率和碱性磷酸酶也可以增加,但是无诊断特异性。
2.血管紧张素转化酶(ACE)40%~90%的活动性结节病患者有血清ACE增高,但由其他疾病造成的假阳性将近20%。
3.可溶性白介素-2受体(soluble interleukin-2 receptor,sIL-2R)sIL-2R反映T细胞的活动,结节病有肺外脏器受累时伴随着血清sIL2R增高,提示血清sIL2R可能是监测结节病活动和病情严重程度的指标。4.结核菌素试验 对PPD5TU的结核菌素皮肤试验无反应是结节病的特点,可以用来鉴别结核和结节病。
诊断
1.有组织学证据的诊断标准 ①相应的临床或胸部X线/胸部CT征象;②组织学检查显示非干酪样肉芽肿;③细菌和真菌检查阴性,除外了其他肉芽肿性疾病。
2.如果无组织学证据,依据下列标准之一,足以建立结节病的临床诊断。
(1)有相应的临床或胸部X线/胸部CT征象,而且BAL检查显示CD4+/CD8+>3.5,结节病的诊断多能成立。
(2)具有双肺门淋巴结肿大、关节炎和结节性红斑三联征表现,伴有发热、不适和肌肉痛,则可以诊断为Löfgren综合征,即急性结节病。
(3)67Ga扫描显示Panda征象(两侧肺门淋巴结和右侧气管旁淋巴结67Ga聚集显像)和Lambda征象(腮腺和泪腺67Ga聚集显像),可以诊断为结节病,但敏感性只有13%~48%。诊断结节病不仅要做到组织/细胞学证实,还要评估脏器累及范围与功能损害程度,判断疾病活性,以决定治疗与否。评价结节病活动性最好的方法是观察活动的情况,包括发作的方式,症状是否恶化或持续存在,皮肤损害的变化,并应结合胸片和肺功能的改变。血清ACE水平、67Ga扫描、胸部HRCT以及BAL中CD4+/CD3+比值也可以用来反映疾病活动的情况。
鉴别诊断
有肺门淋巴结肿大者应该与下列疾病鉴别:
1.肺门淋巴结结核 肺门淋巴结肿大一般为单侧性,有时可钙化,可见肺部原发灶。患者多在20岁以下,常有低度毒性症状,PPD试验多为阳性。
2.淋巴瘤 常见全身症状如发热、消瘦、贫血等,胸内淋巴结肿大多为单侧或双侧不对称肿大。借助组织学检查可以鉴别。
3.肺门转移性肿瘤 肺部和肺外肿瘤转移至肺门或纵隔淋巴结多形成单侧或双侧不对称肿大,通常有原发病灶.借助细胞学和组织学检查可以鉴别。
4.其他肉芽肿性疾病或弥漫性肺实质疾病 如外源性过敏性肺泡炎、铍肺、感染性(结核杆菌和真菌)、化学性因素所致肉芽肿应与结节病进行鉴别,通过临床病史及相关检查可以进行鉴别。晚期形成肺纤维化者还需要与特发性肺纤维化等进行鉴别。
治疗
(一)治疗原则
结节病的自然缓解率在1期是55%~90%,Ⅱ期为40%~70%,Ⅲ期为10%~20%。因此,无症状和肺功能正常的Ⅰ期、Ⅱ期和Ⅲ期结节病患者,一般不需要特殊的治疗,但需要跟踪观察。(二)治疗药物
1.糖皮质激素 结节病采用全身糖皮质激素治疗的适应证包括生命或视力受到威胁的脏器受累,如心脏、中枢神经系统或眼部受累,持续性高钙血症,持续性肾功能不全,严重的肝功能障碍伴门脉高压或黄疸、脾大或脾功能亢进,严重的乏力和消瘦,皮肤损害或慢性肌病。肺结节病如果出现肺部症状,肺功能障碍严重或逐渐恶化,影像学表现加重时,则需要糖皮质激素治疗。通常采用泼尼松,初始剂量0.5mg/(kg·d),连续4周,然后根据治疗反应每4周减量5~10mg,逐渐减量为每天5~10mg后维持治疗,疗程6~24个月。停药后的复发率为16%~74%。对于有复发倾向的患者,应该适当增加激素的剂量。吸入激素可以缓解支气管的症状,但对于肺结节病的治疗价值有限.
2.替代治疗 其他免疫抑制剂或抗炎治疗对于结节病仅有有限的治疗作用,而且副作用比较大,因此只是限于激素治疗无效或有严重副作用的患者。硫唑嘌呤口服剂量为每天100~150mg或甲氨蝶呤口服剂量为每周10~20mg,目前多选择后者。已酮可可碱、英夫利昔单抗(infliximab)等药物治疗的有效性与安全性值得进一步研究证实,目前不推荐常规应用。
3.肺移植 肺移植是终末期肺结节病可以考虑的唯一有效的治疗方法。移植指征是活动耐力下降(NYHa功能Ⅲ或Ⅳ级),符合下列任一条:①静息状态下低氧血症;②肺动脉高压;③右房压增高,大于15mmHg。(三)跟踪观察
结节病Ⅰ期随访的原则是每6个月复查1次,其他期每3~6个月复查1次,治疗停止后随访至少3年,尤其是对糖皮质激素治疗缓解的病人要加强随访复查。如果X线正常化达3年,才可以停止继续随访。严重的肺外病变需要长期随访。
预后
结节病的病程和预后变化很大,自发缓解率为70%,慢性病程者仅占10%~30%,病死率为1%~5%,其中75%的死亡与进展期肺结节病有关,但是目前还没能确定重症慢性进展性结节病的预后判断指标。
外源性过敏性肺泡炎外源性过敏性肺泡炎(extrinsicallergicalveolitis,EAA),也称过敏性肺炎(hypersensitivity pneumonitis),是指易感个体反复吸入有机粉尘抗原后诱发的一种主要通过细胞免疫和体液免疫反应介导的肺部炎症反应性疾病,以淋巴细胞渗出为主的慢性间质性肺炎,细胞性细支气管炎(气道中心炎症)和散在分布的非干酪样坏死性小肉芽肿为特征性病理改变。许多职业暴露可以引起EAA,根据不同的职业接触和病因又有很多具体的疾病命名。农民肺是EAA的典型形式,是由吸入霉干草中的嗜热放线菌或热吸水链霉菌孢子所致。吸入含嗜热放线菌的蘑菇肥料引起蘑菇工人肺,吸入发霉甘蔗粉尘中的嗜热放线菌引起蔗尘肺,吸入含动物蛋白的羽毛和排泄物尘埃引起饲鸟者肺(如鸽子肺、鹦鹉肺),生活在有嗜热放线菌污染的空调或湿化器的环境引起空调器肺等。各种病因所致EAA的临床表现相同,可以是急性、亚急性或慢性。
临床表现
急性形式是最常见和具有特征的表现形式,一般在明确的职业粉尘或环境抗原接触后4~8小时出现畏寒、发热、全身不适伴胸闷、呼吸困难和咳嗽。两肺底部可闻及细湿啰音或细小爆裂音,偶闻哨鸣音。病情轻重与吸入抗原的量与暴露时间有关。如果脱离抗原接触,病情可于24~48小时内恢复。如果持续暴露,则接触和症状发作的关系可能不明显,反复急性发作导致几周或几个月内逐渐出现持续进行性发展的呼吸困难,伴体重减轻,表现为亚急性形式。
慢性形式是长期暴露于抗原导致急性或亚急性疾病反复发作后的结果。主要表现为进行性发展的呼吸困难伴咳嗽和咳痰及体重减轻。肺底部可以闻及吸气末细小爆裂音,少数有杵状指。晚期有发绀、肺动脉高压及右心功能不全征象。
辅助检查
1.X线胸 片急性/亚急性形式主要表现以双侧中下肺野分布为主的斑片状或弥漫性磨玻璃样变,或伴网状或网结节影。也有很多病人的胸片无异常,或仅表现双肺纹理模糊。影像学的变化与症状的关系不明显。慢性形式主要表现以上、中肺野分布为主的结节、粗线条或网状影,疾病晚期还有肺容积减小、纵隔移位以及肺大疱形成。
2.胸部CT/HRCT 急性/亚急性形式主要显示弥漫性分布的边界不清的小结节影,呈小叶中心性和细支气管中心性分布。斑片性磨玻璃样变和肺泡过度充气交错形成马赛克(mosaic)征象。慢性形式主要表现小叶间隔和小叶内间质不规则增厚,蜂窝肺伴牵拉性支气管或细支气管扩张和肺大疱。间或混有斑片性磨玻璃样变。这种改变类似于IPF,不同的是EAA的纤维化一般不影响肋膈角(彩图8,彩图9)。
3.血液化验 外周血白细胞有一过性和轻度增高,血清IgE正常。血清特异性沉淀抗体(IgG)阳性也可见于无症状的抗原接触者,因此这种特异性抗体的存在只说明有过敏原接触史,并无诊断特异性。
4.肺功能试验 疾病早期可能仅表现弥散功能障碍,P(A-E)DO2增加和运动时低氧血症,随着疾病进展出现限制性通气功能障碍。少数病人还有小气道阻塞和气道高反应性。
5.支气管肺泡灌洗 BALF中淋巴细胞明显增加,尤其是CD8+T细胞增加明显,导致CD4+/ CD8+<1或正常。在抗原吸入后的急性反应期,BALF的中性粒细胞的比例可以呈中度增加,表现为一过性的中性粒细胞性肺泡炎。有时也可见肥大细胞增加。
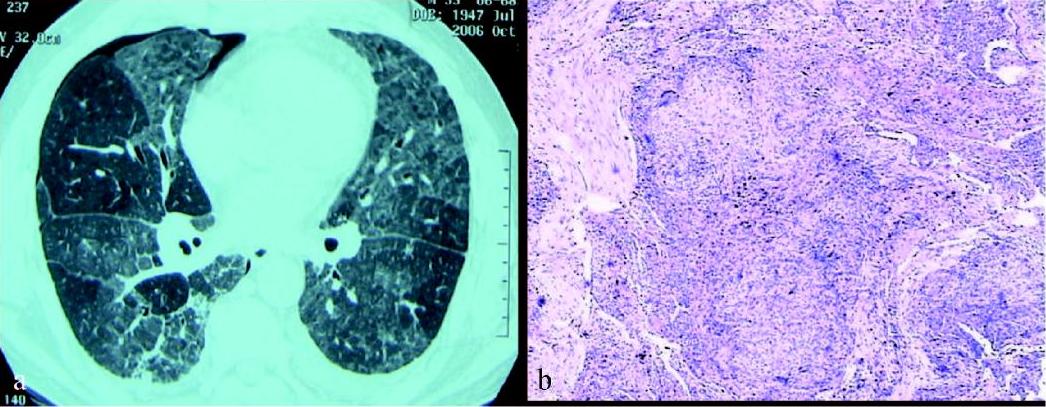
彩图8 女性患者,31岁,活动后呼吸困难1个月,家 里养鸽子20只,诊断亚急性外源性过敏性肺泡炎
彩图9 男性患者,6l岁,嗜养鸽子100只,共10年。出现 活动后气短进行性加重3年,伴少许咳嗽。诊断慢性外源性过敏性 肺泡炎
诊断与鉴别诊断
根据明确的抗原接触史,典型的症状发作特点,胸部影像学和肺功能的特征性改变,BALF检查显示明显增加的淋巴细胞和CD4+/CD8+<1,可以做出明确的诊断。TBLB取得的病理资料能进一步支持诊断,通常不需要开胸肺活检。表8列出了建立外源性过敏性肺泡炎的诊断标准,如果满足4个主要标准和2个次要标准或除外结节病、IPF等,EAA诊断可以确定。

表8 建立外源性过敏性肺泡炎的诊断标准
急性EAA需要与感染性肺炎(病毒、支原体等)鉴别,另外也需要与职业性哮喘鉴别。慢性EAA需要各种其他原因所致的间质性肺炎、结节病和肺结核进行鉴别。
治疗
根本的治疗措施是脱离或避免抗原接触,改善作业卫生、室内通风和空气污染状况,降低职业或环境有机粉尘的吸入。单纯的轻微呼吸道症状在避免抗原接触后可以自发缓解,不必特殊治疗。但对于急性重症和慢性进展的患者则需要使用糖皮质激素,其近期疗效是肯定的,但是其远期疗效还没有能确定。急性重症伴有明显的肺部渗出和低氧血症,经验性使用泼尼松0.5mg/(kg·d),1~2周或直到影像学和肺功能明显改善后减量,疗程4~6周。亚急性减量至10~15mg后,维持治疗,疗程3~6个月。慢性,维持治疗时间可能需要更长。
其他间质性肺疾病结缔组织疾病相关性间质性肺疾病
间质性肺疾病可以并发于任一种结缔组织疾病(connective tissue disease,CTD),但是最常见于类风湿关节炎(rheumatoid arthritis,RA)和系统性硬皮病(systemic sclerosis,SSC)。它们的临床表现、生理学、影像学和组织病理学特征类似于IPF,但是与结缔组织疾病相关的肺纤维化病程相对缓慢。结缔组织疾病相关性间质性肺疾病(CTD-ILD)的活动状态或病程与CTD病的肺外或全身表现没有关系。即使无关节或全身症状,肺部病变也可能进展。TBLB和BALF检查虽然没有特异性,但是有助于证实CTD-ILD的诊断且除外其他疾病。因为对于诊断明确的CTD患者,如果有肺纤维化的典型临床表现和HRCT特征,相应的BALF和TBLB结果可以做出CTD-ILD的诊断,所以很少需要开胸肺活检。CTD-ILD患者伴严重的肺功能损害或临床症状加重,起始应用泼尼松0.5~1mg/(kg·d),逐步减量至5~10mg/d,维持治疗。经常需要联合应用环磷酰胺或硫唑嘌呤或其他免疫抑制剂,以预防疾病进展。
药物诱发的间质性肺疾病
药物诱发的肺损害包括:①慢性间质性肺炎;②过敏反应;③肺嗜酸性粒细胞渗出;④药物诱发的狼疮综合征;⑤弥漫性肺泡出血等。
引起ILD最常见的药物包括博来霉素、环磷酰胺和胺碘酮,其中一些制剂(如博来霉素和胺碘酮)还表现出剂量依赖关系。发病机制包括药物的直接毒性作用以及炎症与修复反应所致的间接损害。诊断依赖于:①药物使用与肺损害发生有严格的时相关系,除外其他可能引起肺脏损害的因素;②停药后肺损害减轻,再次用药后肺损害加重;③可能引起肺损害的药物是唯一使用的药物;④药物性肺损害的特征包括临床、影像和/或组织病理表现是以往这个药物曾经出现并报道过的。根据Irey的这一标准,药物与肺损害之间的关系可以分为肯定,很可能和可能三级。只有少数病例能够完全符合上述药物性肺损害的诊断标准,大多数只能是很可能和可能的病例。
如果及时停药,多数药物性肺损害是可逆的,或病变不再进展;如果肺部损害严重,可以根据其可能的组织病理学改变加用糖皮质激素;可疑的药物一般不提倡试验性再次给药,以免造成肺的进一步损害。
嗜酸粒细胞性肺炎
嗜酸粒细胞性肺炎通常以肺部嗜酸粒细胞浸润伴有或不伴有外周血嗜酸粒细胞增多为特征,是几种具有不同病因和病理学特点的疾病的共同表现。它既可以是已知原因(如某些真菌过敏、药物反应和寄生虫感染)引起的疾病,又可以是原因不明的疾病。按临床过程区分为急性嗜酸粒细胞性肺炎和慢性嗜酸粒细胞性肺炎,后者多局限于肺,但是和肉芽肿性血管炎相关的慢性嗜酸粒细胞性肺炎如Churg-Strass综合征常常累及多个组织器官。虽然关于嗜酸粒细胞性肺炎尚没有统一的分类,但是目前倾向于按病因分类,这样有利于因病施治(表9)。
(一)吕弗勒综合征
吕弗勒综合征(Löffler’s syndrome)又称单纯性肺嗜酸粒细胞增多症。其病因及发病机制尚不完有全清楚,目前认为主要与寄生虫感染和药物过敏有关。表现为轻微的呼吸道和全身症状,外周血嗜酸粒细胞增多,胸部X线呈游走性浸润影。本病具有自限性,一般在1个月内可自愈。治疗上主要是去除病因,如驱虫,停用可疑过敏药物,对于症状明显或反复发作者可考虑应用糖皮质激素。

表9 嗜酸粒细胞性肺炎的分类
(二)热带型肺嗜酸粒细胞增多症
热带型肺嗜酸粒细胞增多症(tropical pulmonary eosinophilia)是丝虫感染后,微丝蚴寄宿在肺血管部位时刺激局部发生的过敏反应。因此,本病主要发生于丝虫病流行地区(如印度、斯里兰卡、东南亚、我国的江南地带等)。临床以持续的哮喘和咳嗽为主要症状,并于夜间加重。其他症状包括胸痛、咯血及全身症状如发热、无力和体重减轻。体征包括喘鸣音和/或爆裂音及一些肺外表现(如肝脾大、淋巴结肿大、胃肠道症状、肌肉疼痛、皮肤病变等)。外周血嗜酸粒细胞明显增多(大于2×109/L),血清IgE增高和抗丝虫抗体的滴度增高。X线胸片显示以双侧中下肺野分布为主的斑点状和网格结节样渗出影,边界不清,相互融合。肺功能主要表现为限制性通气功能障碍,气道阻塞不明显。治疗首选抗丝虫药乙胺嗪(海群生),按6~8mg/(kg·d),分3次口服,治疗10~14天,症状通常于治疗2周内迅速消失。复发患者对再次治疗仍然敏感,但是疾病长期不愈可能导致不可逆的肺纤维化和病程迁延。
(三)变应性支气管肺曲菌病
变应性支气管肺曲菌病(allergic bronchopulmonaryaspergillosis,ABPA)是嗜酸粒细胞性肺炎中比较常见的一种,主要是烟曲菌(也包括其他曲菌和念珠菌)刺激易感个体后,通过产生Ⅰ和Ⅲ型免疫反应而导致发病。病理改变特点为肺组织中有明显的嗜酸粒细胞渗出,支气管黏液样痰栓及支气管中心性肉芽肿形成导致支气管阻塞或支气管壁的进行性损害,从而出现近端支气管扩张。ABPA多发于中年女性,呈反复发作的哮喘伴咳嗽,咳出特征性的稠硬痰块或痰栓后症状缓解。胸片和胸部CT主要表现双侧肺部多发斑片或片状渗出影,呈游走性改变;支气管痰栓阻塞导致近端支气管扩张,阻塞远端出现肺不张和阻塞性肺炎,而形成“V”或“Y”型或葡萄状阴影。外周血嗜酸性粒细胞通常呈现中等度增高,达20%~30%或(0.5~2)×.109/L,也可更高。痰中嗜酸粒细胞增高和曲菌或念珠菌阳性,血清IgE明显增高(>2000ng/ml),伴IgG增高。90%病例的血清曲菌沉淀抗体阳性。曲菌皮肤针刺试验阳性。
诊断ABPA的主要条件包括哮喘、外周血嗜酸性粒细胞增多、肺部浸润影、血清曲菌特异性IgE或曲菌皮肤针刺试验阳性。次要条件包括血清曲菌特异性沉淀抗体阳性、血清总IgE增高和近端支气管扩张。
ABPA的基本治疗是使用糖皮质激素。一般采用口服泼尼松,起始剂量为0.5mg/(kg·d),当临床症状缓解,肺部阴影消失,血嗜酸粒细胞降低或血清IgE降低后,逐渐减量,疗程3~6个月。其他治疗包括控制哮喘和溶解清除痰栓治疗。抗真菌治疗(如伊曲康唑)的临床价值有待验证。
(四)慢性嗜酸粒细胞性肺炎
慢性嗜酸粒细胞性肺炎(CEP的发病原因不明,最常发生于中年女性,通常于数周或数月内出现呼吸困难、咳嗽、发热、盗汗、体重减轻和喘鸣,呈现亚急性或慢性病程。也有表现为迅速进展到严重呼吸衰竭的急性形式(急性嗜酸粒细胞性肺炎)。
X线胸片的典型表现有肺外带(胸膜下)的致密肺泡渗出影,中心带清晰,这种表现称作“肺水肿反转形状”(photographic negativeof pulmonary edema),而且渗出性病变多位于上叶。80%的病人有外周血嗜酸粒细胞增多。血清IgE增高也常见。典型的肺功能改变为限制性通气功能障碍,经常伴气流阻塞、DLco降低和低氧血症。
如果患者有相应的临床和影像学特征,BALF嗜酸粒细胞大于40%,高度提示嗜酸粒细胞性肺炎。很少需要外科肺活检,因为根据症状、特征性的影像学改变以及BALF和TBLB可以做出诊断,但需除外其他原因引起的肺部嗜酸粒细胞增多。
治疗首选糖皮质激素,通常应用泼尼松,初始剂量0.5~1mg/(kg·d),连续2~4周后,依据治疗反应逐渐减量至10mg/d,维持治疗,总疗程6~12个月。具有复发倾向的病人可能需要使用小剂量泼尼松长期维持治疗。
硅沉着病
硅沉着病(silicosis)过去称矽肺,是由于长期吸入大量游离二氧化硅所引起的弥漫性结节性纤维化性肺疾病,它是尘肺中最常见、进展最快、危害最严重的一种类型。主要表现隐匿性进行性发展的呼吸困难伴咳嗽,限制性通气功能障碍和弥散功能障碍。X线胸片是诊断矽肺的主要方法,特征性地显示结节阴影(直径1~3mm)、网状阴影或/和大片融合病灶。其次为肺门改变、肺纹理改变和胸膜改变。因肺纤维化收缩使肺门上移,增粗的肺纹理呈垂柳状,并出现气管纵隔移位。肺门阴影密度增加,有时可见“蛋壳样钙化”的淋巴结。胸膜可以增厚、粘连或钙化。
根据患者详细的职业史和密切的二氧化硅粉尘接触史,结合相应的临床表现和X线胸片特点可以做出诊断。硅沉着病需要与急性粟粒性肺结核、结节病、肺泡癌、肺含铁血黄素沉着症以及肺泡微石症等相鉴别。
处理对策在于全面实行三级预防措施,做到及时预防、早期检测、早期处理、合理补救。目前尚无逆转纤维化病变的药物,多采用对症支持治疗以减轻症状。
罕见间质性肺疾病
(一)肺朗格汉斯细胞组织细胞增生症
成人的朗格汉斯细胞组织细胞增生症通常局限于肺,因此称作肺朗格汉斯细胞组织细胞增生症(PLCH)。病变以呈细支气管中心分布的朗格汉斯细胞渗出形成的肉芽肿性改变,并机化形成“星形”纤维化病灶,伴囊腔形成为病理改变特征。PLCH罕见,主要见于吸烟人群,常见症状是咳嗽和呼吸困难。X线胸片显示结节或网格结节样渗出性病变,常分布于肺上叶和中叶,肋膈角清晰,进展病例可见明显的蜂窝肺、纤维囊性变和气胸。HRCT特征性地表现多发的薄壁囊腔,囊腔可以融合形成直径超过2~3cm的肺大疱,细支气管周围结节(直径1~4mm)。主要涉及上叶肺的多发性囊腔和结节或BALF朗格汉斯细胞(OKT6或anti-CD1a抗体染色阳性)超过5%高度提示PLCH的诊断。确定诊断需要肺组织活检,外科肺活检优于TBLB。
因为疾病可能是自限性的,病程难以预计,2/3以上的病人在发病6~24个月内,病情稳定或改善。主要治疗是首先劝告病人戒烟。对于严重或进行性加重的病人,尽管已经戒烟,还需要应用糖皮质激素。
(二)肺淋巴管平滑肌瘤病
肺淋巴管平滑肌瘤病(PLAM)几乎只发生于育龄妇女,发病原因不明。病理学以肺泡壁、细支气管壁和血管壁的类平滑肌细胞(LAM细胞,HMB-45+)呈弥漫性或结节性增生,导致局限性肺气肿或薄壁囊腔形成,最终导致广泛的蜂窝肺为特征。
临床上主要表现进行性加重的呼吸困难、咯血、反复出现的气胸和乳糜胸。肺功能呈现气流阻塞和气体交换障碍,有时伴有限制性通气功能障碍。典型的胸片是网格和囊腔样变。胸部HRCT特征性地显示薄壁囊腔(直径2~20mm)弥漫性分布于两侧肺。早期可能只有稀疏分布的囊腔,随疾病进展,囊腔数目增多,并融合形成不规则囊腔。LAM与PLCH在CT上的主要区别是PLCH一般不影响肋膈角,囊腔壁更厚,疾病早期有更多的结节。
对于PLAM尚无有效的治疗方法。目前临床上还在使用的孕激素治疗并没有研究证实有效。近来研究显示免疫抑制剂西罗莫司(雷帕霉素)具有潜在的治疗效果,但还需要多中心临床试验验证。终末期PLAM可以考虑肺移植。
(三)肺泡蛋白沉积症
肺泡蛋白沉积症(PAP)以肺泡腔内积聚大量的磷脂蛋白、类表面活性物质为特征。通常表现隐匿性呼吸困难和咳嗽,偶有咳痰。肺部闻及吸气性爆裂音。X线胸片显示两侧弥漫性的肺泡渗出,分布于肺门周围,形成“蝴蝶”样图案。经常是广泛的肺部渗出与轻微的临床症状不相符合。胸部HRCT特征性地表现:①磨玻璃影与正常肺组织截然分开,形成“地图”样图案;②小叶间隔和小叶内间隔增厚,形成多边形或“不规则石块面路”样图案;③大片实变影,伴支气管气像,实变周围是磨玻璃影。特征性生理功能改变是肺内分流导致的严重低氧血症。80%的病人有血清乳酸脱氢酶增高。BALE回收液特征性地表现奶白色,稠厚且不透明,静置后沉淀分层,BALF细胞或TBLB组织的过碘酸雪夫(PAS)染色阳性和阿辛蓝染色阴性可以证实诊断。
1/3的病人可以自行缓解。对于有明显呼吸功能障碍的患者,全肺灌洗是首选和有效的治疗。全肺灌洗可使75%~95%患者的病情改善,但约1/3可复发,需要再次灌洗。(四)特发性肺含铁血黄素沉着症
特发性肺含铁血黄素沉着症(IPH)的发病原因不明,多发生于儿童和青少年,以反复发作的弥漫性肺泡内出血,导致咯血、呼吸困难和缺铁性贫血为临床特点。胸部X线的典型表现是两肺中、下肺野弥漫性分布的边缘不清的斑点状阴影。当肺内出血停止,肺泡渗出影吸收并演变成弥漫性网织结节影。过程反复导致肺纤维化形成。大约半数病人的外周血嗜酸性粒细胞轻度增高和冷凝集试验阳性。
诊断主要根据发复的咯血、肺内弥漫分布的边缘不清的斑点状阴影及继发的缺铁性贫血做出初步诊断。常规进行BaL检查可确诊有无肺泡出血,尤其是对有无隐匿性出血。BALF检查发现游离红细胞或含吞噬红细胞的肺泡巨噬细胞提示近期肺泡出血,查找到许多含铁血黄素巨噬细胞提示远期肺泡出血。同时也应该常规检测循环自身免疫抗体(如anti-GBM、ANCA、ANA、RF等)以除外其他原因所致的弥漫性肺泡内出血。
一般而言,IPH的临床过程比较轻,尤其在成年人,25%可以自行缓解。但是广泛肺泡内出血也可导致死亡。治疗以支持治疗为主。糖皮质激素联合硫唑嘌呤或环磷酰胺治疗对于改善急性加重期的预后和预防反复出血有益,但是尚无确定的疗效判断指征。
处置建议
治疗原则明确病因最为关键,与结缔组织病、药物、毒物、感染相关者应针对病因治疗
特发性间质性肺病的治疗和预后取决于患者的临床-影像-病理诊断(CRP诊断)
已诊断结缔组织病患者出现呼吸症状需鉴别结缔组织病相关肺间质病和肺部感染
病情评估/分型对于原因不明的间质性肺炎,即特发性间质性肺炎(IIP),主要分为三种类型:主要的IIP(包括IPF、INSIP、DIP、RB-ILD、COP和AIP)、罕见的IIP(包括ILIP、IPPF)及不能分类的IIP。确诊依赖于多学科讨论的CRP诊断
一般治疗消除可能的原因,如避免药物、职业接触史,RB-ILD应戒烟
IPF患者,静息低氧血症患者需要接受长期氧疗,多数呼吸衰竭时不应该接受机械通气
药物治疗可复性类型但具有进展危险(细胞型非NSIP、DIP、COP),需要长期合理用药治疗
IPF尚无特异性治疗药物
合并症治疗多数IPF患者,不应该接受针对肺动脉高压的治疗,对于无症状胃食管反流,多数患者应该接受治疗
其他治疗某些合适的IPF患者,应该接受肺移植治疗
康复治疗IPF患者,多数应该进行肺康复
姑息治疗应减轻IPF患者生理和心理上的痛苦
用药建议
结缔组织病相关肺间质病已诊断结缔组织病患者有呼吸症状需除外肺部感染
对糖皮质激素(CS)和免疫抑制剂的疗效优于IPF
剂量需个体化,根据结缔组织病活动性及时调整 剂量,避免患者免疫功能下降,诱发严重感染
治疗目标:以最小剂量控制疾病进展
特发性肺纤维化无治疗有效药物,不应该接受CS单药治疗,不应该接受秋水仙碱、环孢素A、干扰素-γ、益普赛、CS联合免疫抑制剂治疗
多数IPF 患者不应该接受,少数可能是合理选择,包括N-乙酰半胱氨酸、吡非尼酮、抗凝治疗
隐源性机化性肺炎CS:迅速改善症状,病灶吸收可不遗留瘢痕
方案:泼尼松每日0.75-1.5mg/kg,维持4-6周后逐渐减量,疗程1年。或甲基强的松龙每日500-1000mg 冲击,后每日20mg维持
复发后应用CS仍然有效
硫唑嘌呤、环磷酰胺(CTX)、环孢素:一般不需要使用,激素不良反应非常明显者,可应用以减少激素剂量
非特异性间质性肺炎CS:为首选用药。强调剂量个体化,绝大多数症状可改善甚至完全缓解
无CS使用禁忌者,泼尼松每日40-60mg或1mg/kg,根据治疗反应减量,一般1-3月后减至20-40mg/d,4-6月后减至10-15mg/d。总疗程1年
免疫抑制剂:以纤维化表现为主型、CS不能耐受或治疗效果不佳者,可以低剂量CS联合硫唑嘌呤或CTX治疗
非特异性间质性肺炎(NSIP)细胞为主型NSIP疗效优于纤维化型
检查建议
影像学胸部高分辨CT(HRCT),分辨率高,可发现早期肺泡和间质病变。可见毛玻璃影、网结影、结节影、蜂窝肺等。诊断普通型间质性肺炎(UIP)阳性预测值90%以上,足以诊断特发性肺纤维化(IPF)
常规CT扫描和X线胸片可以正常
镓(67Ga)扫描,显示肺泡炎,无特异性,不能预测激素治疗反应和预后
同位素通气和血流扫描:显示通气和血流分步不均,比例失调
X线胸片:有助于评估病变分布,鉴别诊断
肺功能典型为限制性通气功能障碍,弥散功能减低
支气管镜检查支气管肺泡灌洗液(BALF):白细胞分类对诊断、鉴别诊断和观察疗效都有一定意义,同时进行病原体检查
肺组织病理活检支气管镜下肺活检即TBLB,标本量少,常不能明确诊断
开胸或经外科胸腔镜肺活检,确诊率92%。活检应多个肺叶取样,包括胸膜下肺实质。选择肉眼异常区域的边缘,保留肉眼正常肺组织。
需根据患者实际情况决定是否肺活检。非高龄,相对为早期病变,需要与其他特发性间质性肺炎鉴别,有激素治疗指征,心肺功能可胜任手术者可考虑肺活检
对于经病史、症状、影像学和辅助检查可以诊断者,不需进行肺活检,包括过敏性肺泡炎、药物性肺损害、胶原血管病相关间质性肺病等
血液检查辅助诊断和鉴别诊断。包括血常规和白细胞分类、血生化、血ACE、ANCA、ANA谱、类风湿因子、抗基底膜抗体等
血气分析可见低氧血症,肺泡-动脉血氧分压差增大
6分钟步行试验(6MWT)6MWT期间初见低氧血症,是IPF死亡率增高的一个标志,步行距离较短和结束后心率恢复慢与死亡风险增高相关
并发症评价超声心动图:评价肺动脉高压
监测疾病进展监测呼吸困难或咳嗽等症状、静息和活动状态下的血氧饱和度、肺功能指标、HRCT
IPF患者每3-6月对严重程度进行评估,常规监测FVC和DLCO
患者指导
预防避免接触石棉、木材、金属等毒物和职业暴露、损伤肺组织的药物,不吸烟
随访大多数间质性肺病需要长期随访,评估病情发展
对于可复性和自限性类型IIP,如RB-ILD,需要短期随访(3-6月)评价疗效
IPF患者,建议每3-6个月评估疾病严重程度,监测FVC、DLCO、静息和活动状态下血氧饱和度。心肺运动不推荐作为常规监测病情
结缔组织病相关肺间质病的预后因原发病不同而不完全相同
诊断依据
诊断依据1. 劳力性呼吸困难
2. 影像表现为双侧弥漫性间质性浸润
3. 限制性通气功能障碍及弥散功能下降
4. 组织病理特征为肺间质炎症和纤维化表现
1. 环境、职业相关的间质性肺病
2. 药物、治疗相关的间质性肺病
3. 肺感染相关的间质性肺病
4. 慢性心脏疾病相关的间质性肺病
5. 肺血管炎及结缔组织病相关的间质性肺病
6. 肝病、肠道相关的间质性肺病
7. 特发性间质性肺炎
8. 其他病因间质性肺病:如结节病、肺淋巴管平滑肌瘤病、肺泡蛋白沉积症等
1. 除外其他已知病因所致的间质性肺疾病,如职业接触、室内外环境暴露、结缔组织病和药物性肺损害等
2. 未行外科肺活检的患者,HRCT表现为UIP型
3. 行外科肺活检的患者,结合HRCT和外科肺活检符合特定的类型




